Francesco Brunetti,Anna Paola Lugli
Istituto di Dermatologia, Università Cattolica del Sacro Cuore-Fondazione Policlinico Universitario A. Gemelli IRCCS, Roma, Italia
Caso clinico: Si riporta il caso di una paziente di 30 anni, inviata alla nostra attenzione dai colleghi della gastroenterologia, per la comparsa di multiple lesioni a livello degli arti inferiori.
La paziente è affetta dal 2008 da pancolite ulcerosa,in trattamento dal 2015 con adalimumab alla dose di 40 mg ogni 7 giorni e mesalazina (1200 mg 2 compresse al giorno) al momento dell’osservazione
La paziente riferisce l’insorgenza, da circa due mesi, di multiple lesioni papulose eritematose, non pruriginose, con evoluzione cicatriziale, localizzate esclusivamente a livello degli arti inferiori (Figura 1).
All’esame clinico erano apprezzabili, a livello di entrambe le gambe ,isolate lesioni papulose, millimetriche, con atrofia centrale ed eritema periferico; lesioni compatibili con papulosi atrofizzante di Degos.
Con questo sospetto clinico è stata eseguita una biopsia cutanea a livello di una delle lesioni (Figura 2) con riscontro atrofia epidermica, trombosi arteriolare ed ischemia dermica con scarso infiltrato infiammatorio linfo-istiocitario perivascolare ai margini dell’area ischemica.
Il reperto istopatologico supporta la diagnosi di papulosi atrofizzante.
Discussione
La malattia di Köhlmeier-Degos (Papulosi atrofizzante maligna) è una patologia rara caratterizzata da una vasculite trombotica dei piccoli vasi, responsabile di lesioni ischemiche a livello di cute, e, nella forma sistemica, del tratto gastro-intestinale, sistema nervoso centrale e, più raramente, altri visceri.
Le lesioni cutanee papulari con un’atrofia centrale porcellanacea e un alone eritematoso sono considerate patognomoniche [1].
In letteratura sono state individuate due forme di questa patologia:
- Una forma puramente cutanea, benigna, più frequente,
- Una forma sistemica maligna a prognosi temibile [2].
La patogenesi non è nota; alcune segnalazioni di papulosi atrofizzante in più membri di una stessa famiglia suggeriscono la possibilità di una predisposizione genetica [3]. Le principali ipotesi eziopatogenetiche includono una vasculite,una coagulopatia e/o una disfunzione primaria delle cellule endoteliali [1].
Le lesioni cutanee appaiono inizialmente come piccole papule eritematose (da 2,0 a 5,0 mm) sul tronco o sulle estremità, divengono rapidamente necrotiche e sclerotiche, di colore porcellanaceo, circondate da un sottile bordo eritematoso. Nei soggetti che sviluppano un coinvolgimento extra cutaneo i micro-infarti possono interessare qualsiasi tratto del tubo digerente, ma anche altri organi e apparati. I pazienti possono presentare emorragie gastrointestinali, dolori addominali, alterazioni neurologiche e visive, sintomi respiratosi o cardiaci [1-2].
La diagnosi è clinica ma supportata dall’esame istopatologico. Non esistono marcatori sierici specifici, anche se molti pazienti presentano difetti della coagulazione [1].
Non esiste un trattamento efficace. I farmaci che facilitano la circolazione sanguigna (acido acetilsalicilico, pentossifillina, dipiridamolo, ticlopidina, eparina) hanno ottenuto una parziale regressione delle lesioni cutanee in singoli casi e rappresentano un ragionevole approccio terapeutico. L’eculizumab, inibitore della porzione terminale della cascata del complemento, ha determinato un iniziale miglioramento delle lesioni cutanee e intestinali ma non ha impedito la progressione della malattia sistemica. Il treprostinil, analogo della prostaciclina, ha avuto successo in un caso di malattia di Degos, cutanea e intestinale, resistente all’eculizumab. [1]
Bibliografia
- Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Köhlmeier-Degos disease) – a review. Orphanet J Rare Dis. 2013 Jan 14;8:10. doi: 10.1186/1750-1172-8-10. PMID: 23316694; PMCID: PMC3566938.
- A. Theodoridis, A. Konstantinidou, E. Makrantonaki, C.C. Zouboulis, Malignant and benign forms of atrophic papulosis (Köhlmeier–Degos disease): systemic involvement determines the prognosis, British Journal of Dermatology, Volume 170, Issue 1, 1 January 2014, Pages 110–115, https://doi.org/10.1111/bjd.12642
- L. Habbema, L.S. Kisch, Th.M. Starink, Familial malignant atrophic papulosis (Degos’ disease)—additional evidence for heredity and a benign course, British Journal of Dermatology, Volume 114, Issue 1, 1 January 1986, Pages 134–135, https://doi.org/10.1111/j.1365-2133.1986.tb02789.x

Fig.1 – Malattia di Degos: lesioni papulose con atrofia centrale ed eritema teleangectasico periferico.
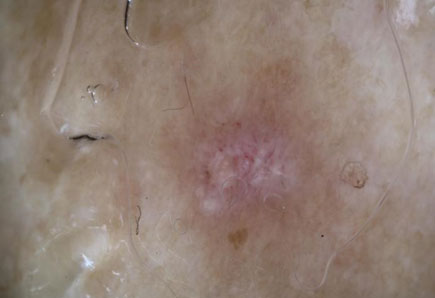
Fig. 2 – Particolare in dermatoscopia delle lesioni cutanee con centro atrofico e alone eritematoso periferico.